
Paper
Biocatalytic sulfation of aromatic and aliphatic alcohols catalyzed by arylsulfate sulfotransferases
I. Oroz‑Guinea, M. Rath, I. Tischler, K. Ditrich, D. Schachtschabel, M. Breuer, W. Kroutil
Appl. Microbiol. Biotechnol. 2024, 108, 520 (17 pages)
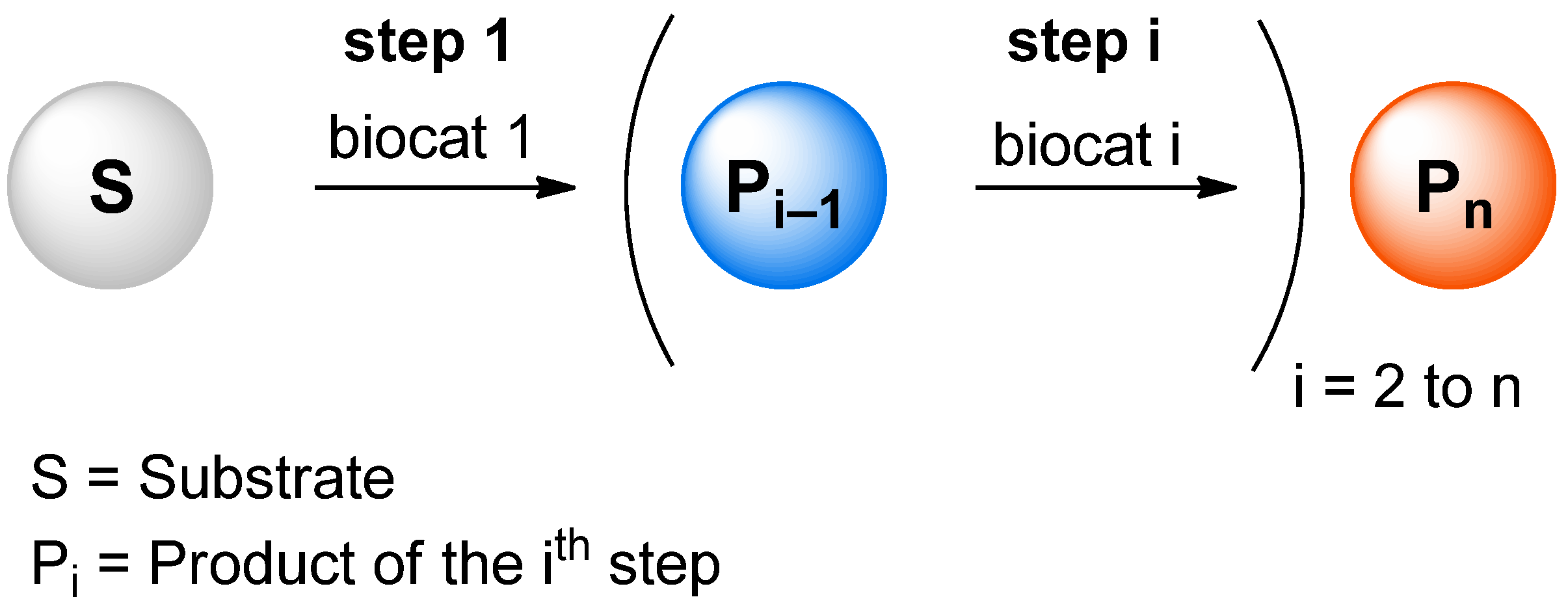
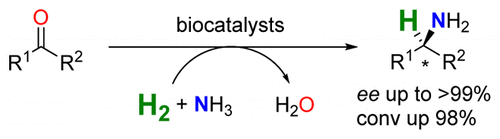
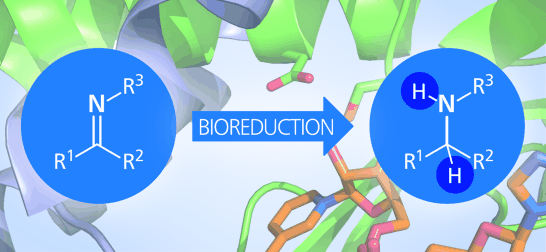
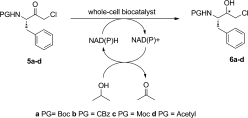
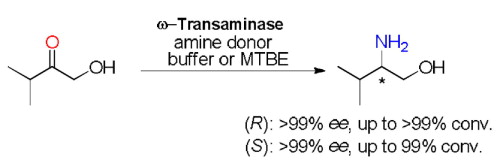
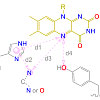
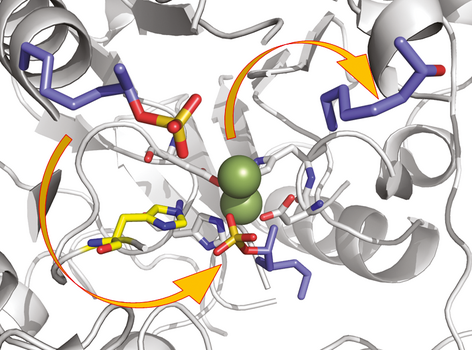
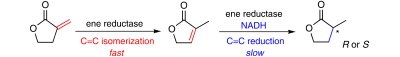
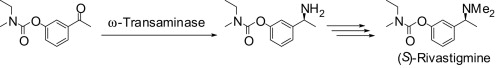
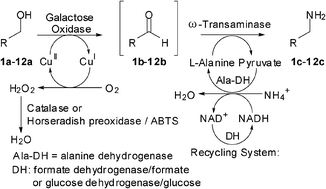
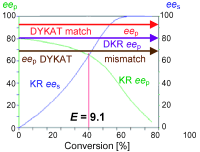
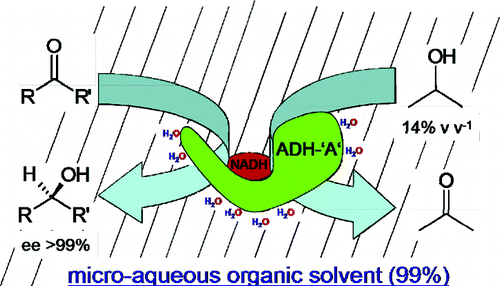
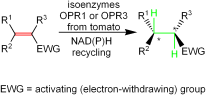
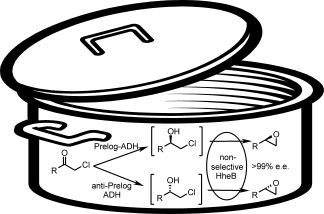
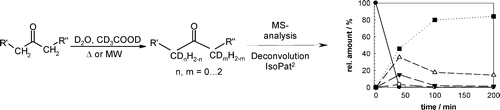
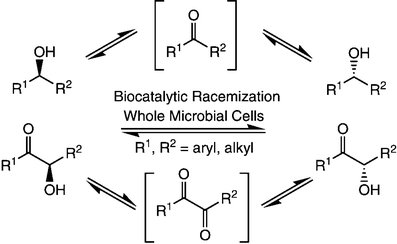
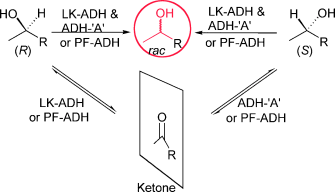
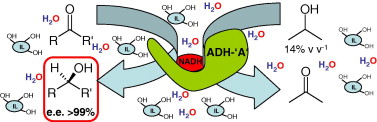
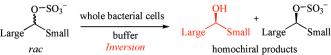
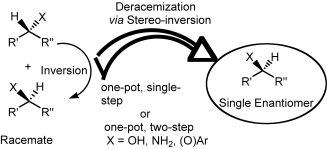
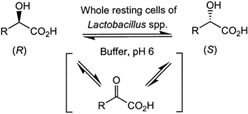
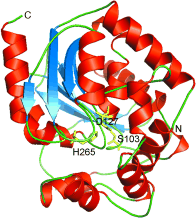
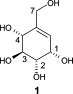
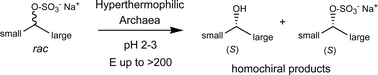
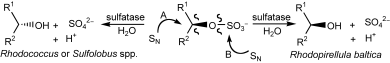
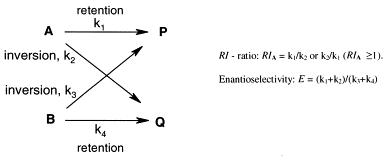
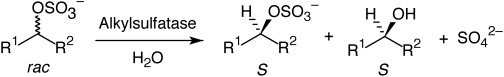
Many relevant metabolites, as well as chemical commodities, contain at least one sulfate ester group. Consequently, biocatalytic strategies to attach sulfate to a molecule under mild conditions are of high interest. In order to expand the enzymatic toolbox available, five new arylsulfate sulfotransferases (ASSTs) were identified in this study. Overexpression in Escherichia coli and enzyme purification resulted in soluble proteins which catalyzed the sulfate transfer to an acceptor substrate using p-nitrophenyl sulfate (pNPS) as sulfate donor. Optimal reaction conditions were established with respect to temperature and pH, as well as their tolerance to organic co-solvents and melting temperature. Additionally, the kinetic parameters (Vmax, KM, and kcat) were determined. The substrate scope for the acceptor showed that a structurally diverse spectrum of alcohols is accepted. The substrates included phenolic alcohols with one, two, and three hydroxy groups, linear and cyclic aliphatic alcohols, and amines. The phenolic substrates were accepted reaching activities of up to 154 U/mg purified enzyme. Additionally, also the aliphatic alcohols (both linear and cyclic) were accepted at reduced activity, showing that these enzymes are not limited to phenolic alcohols. Moreover, catalytic activity was detected when using aniline as an acceptor substrate implying their ability to sulfate also amino groups. Finally, the consecutive sulfation of di- and trihydroxy compounds was observed, resulting in the detection of the corresponding disulfated molecules. KEY POINTS: • Five novel arylsulfate sulfotransferases were identified and characterized. • Accepted substrates included aromatic and aliphatic alcohols, as well as aniline. • Disulfation of di- and trihydroxy aromatic compounds was studied and confirmed.
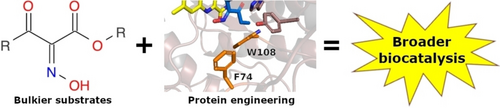
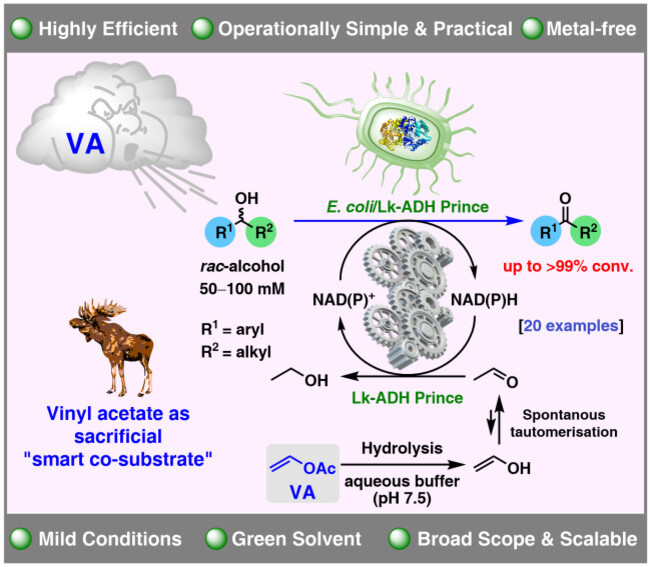
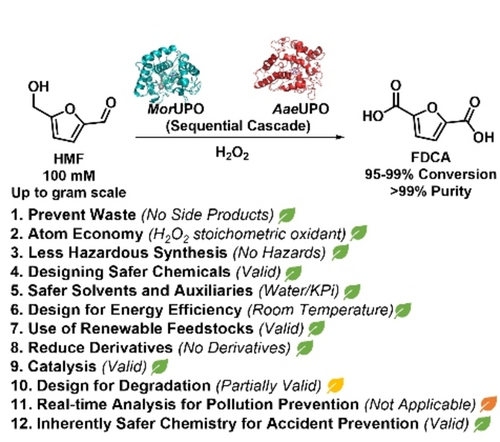
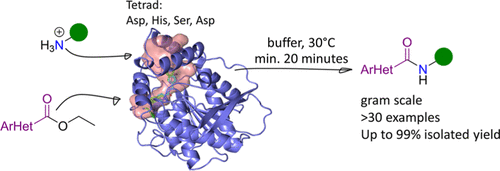
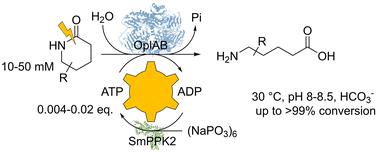
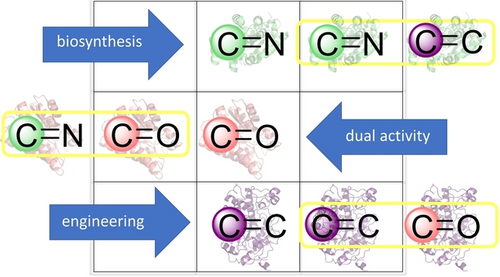
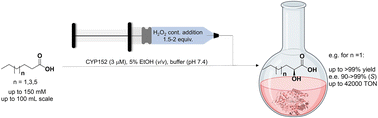
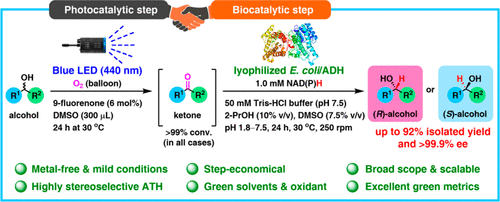
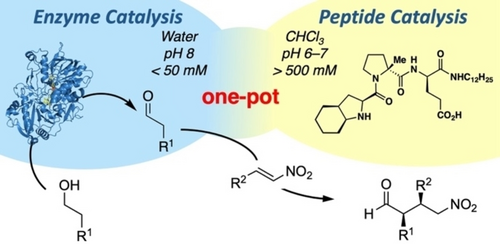
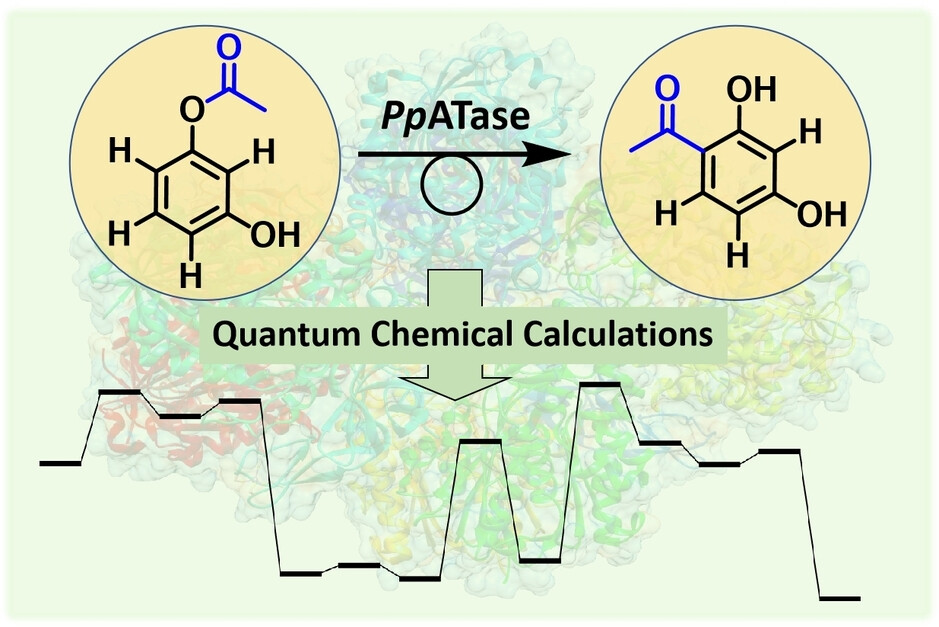
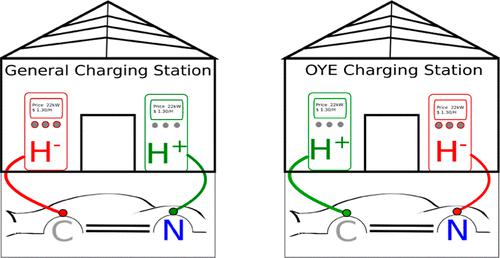
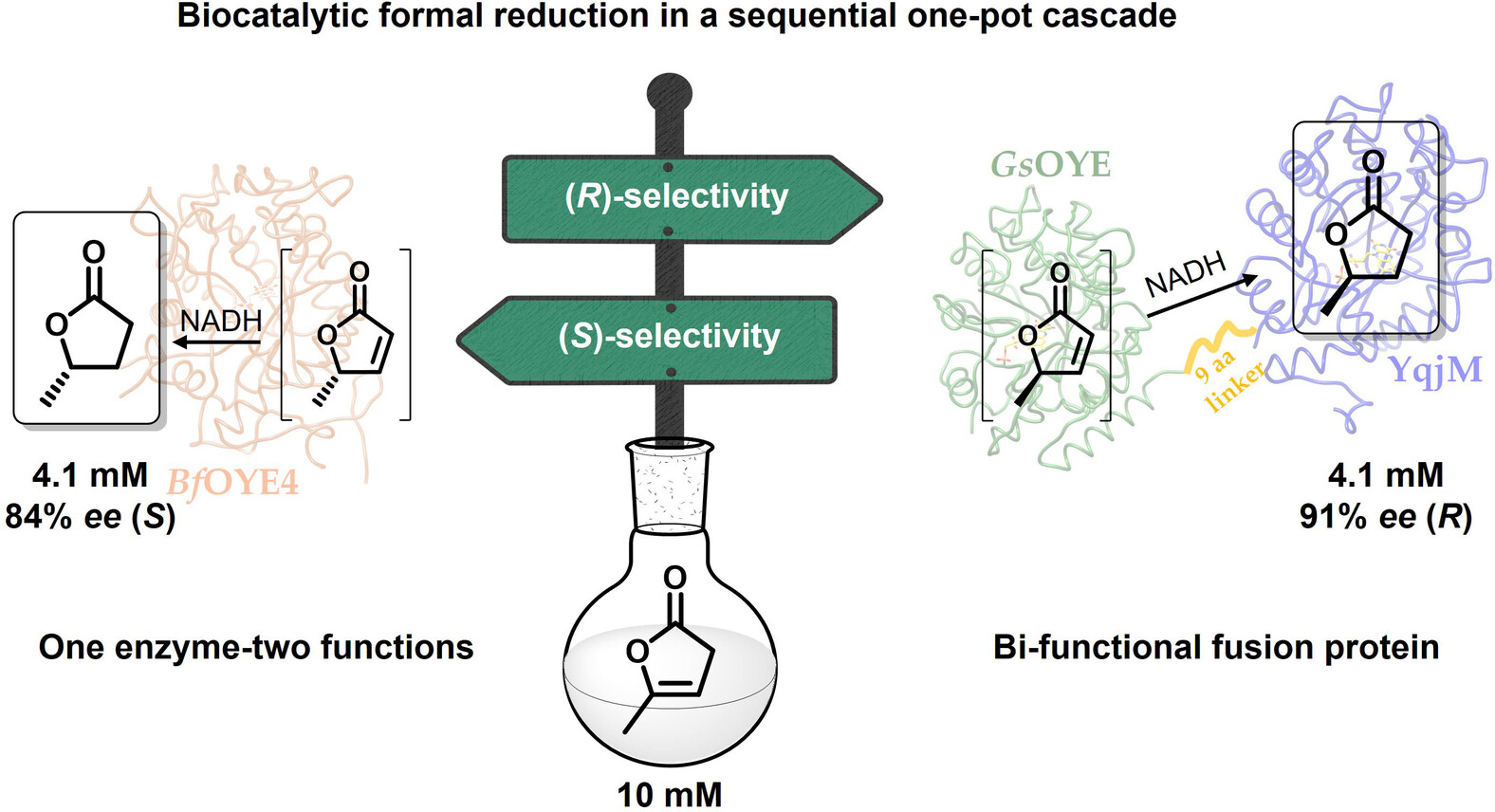





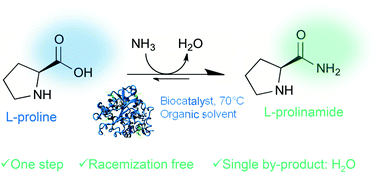
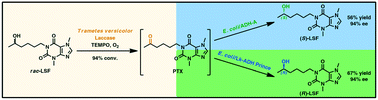
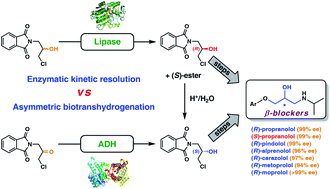
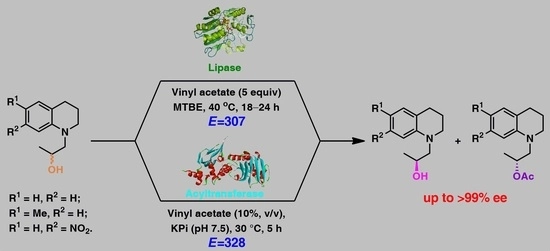
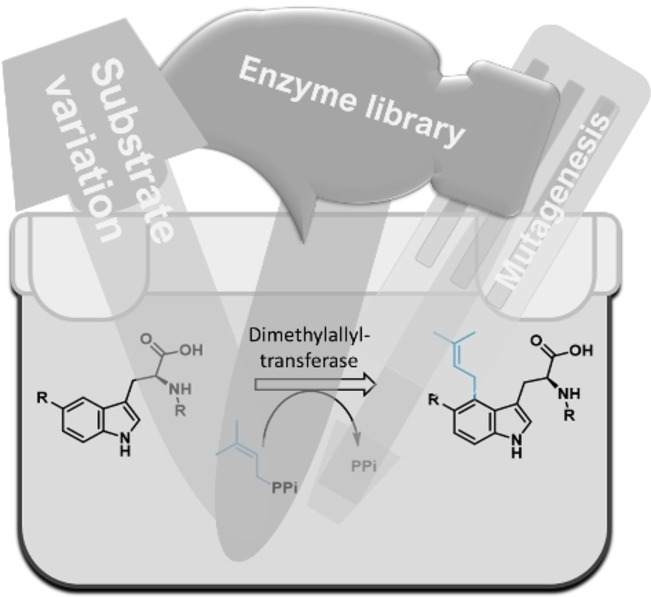
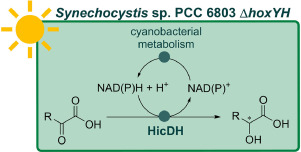
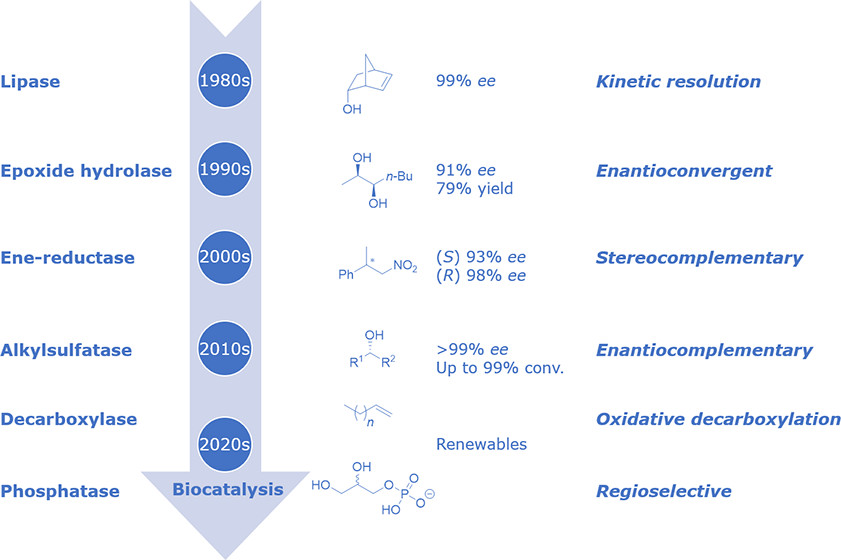
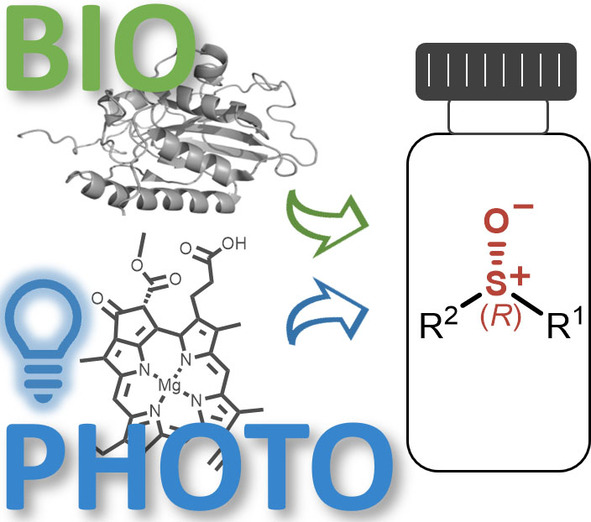


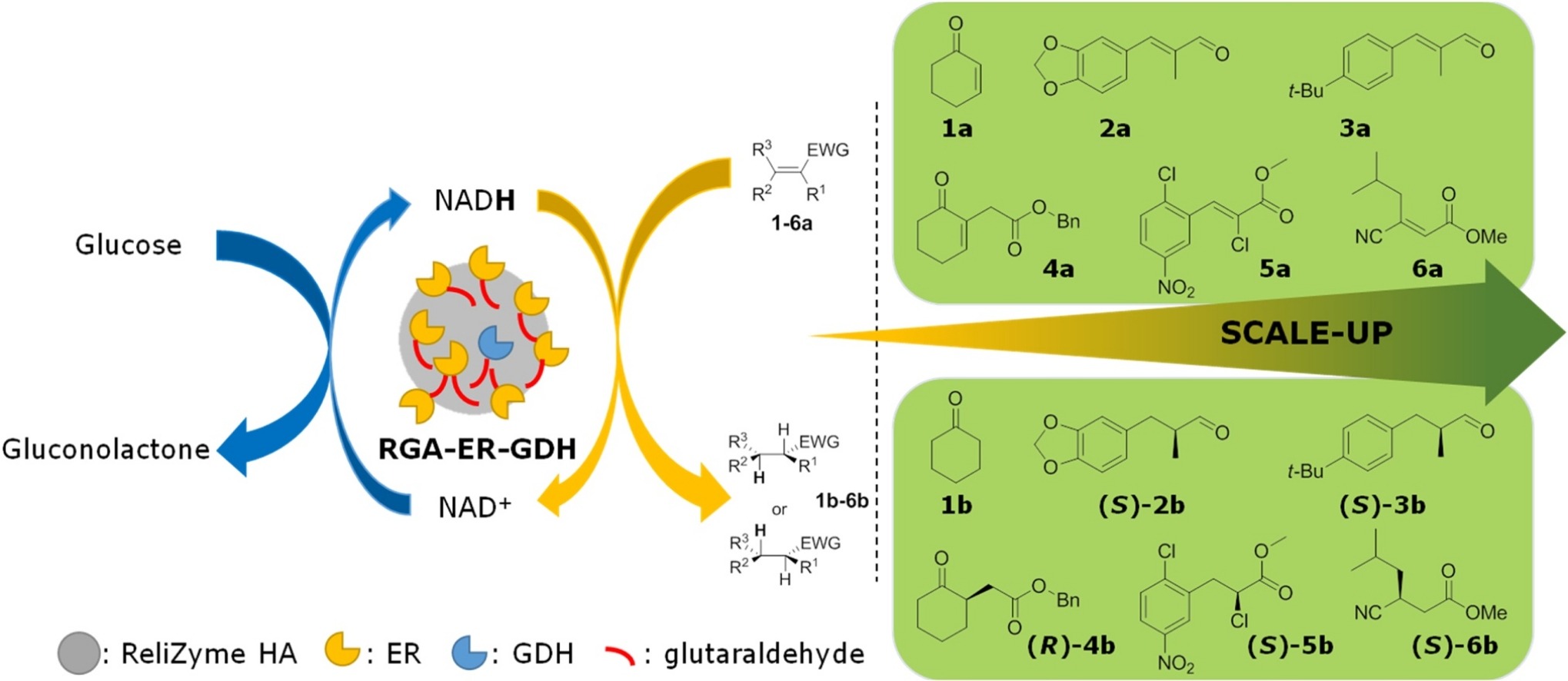
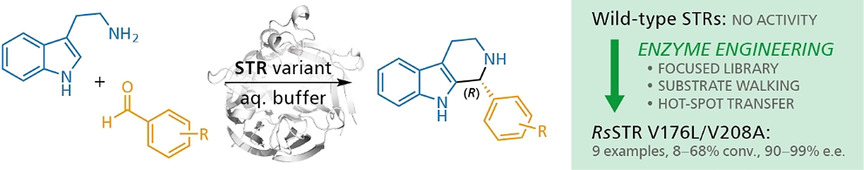
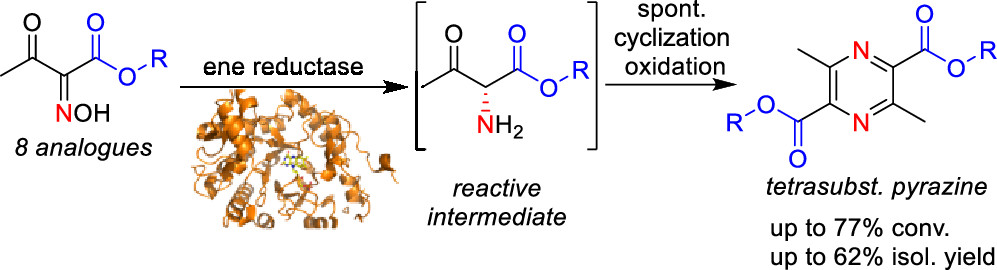
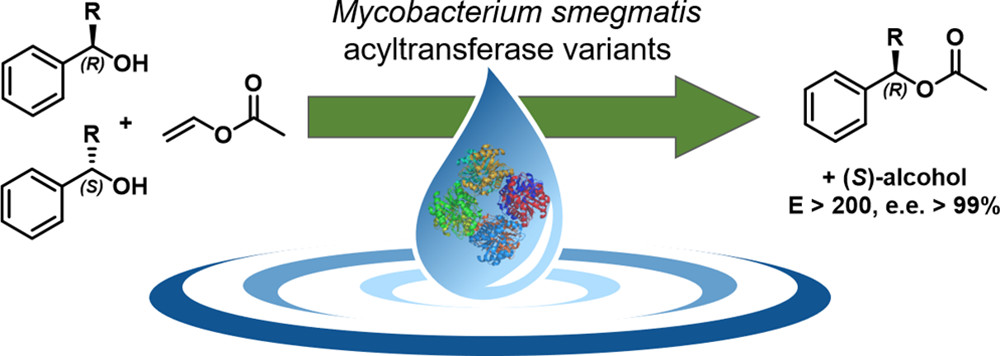
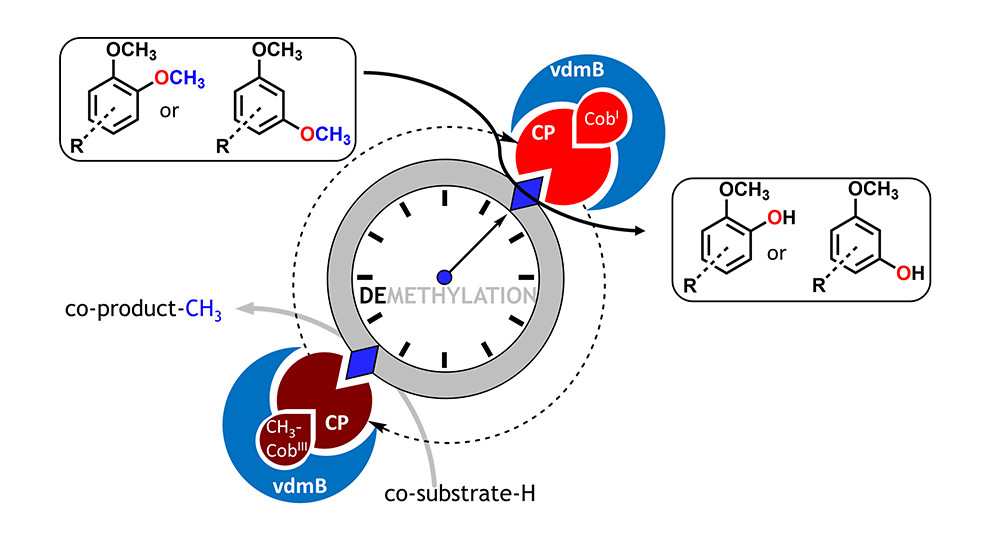
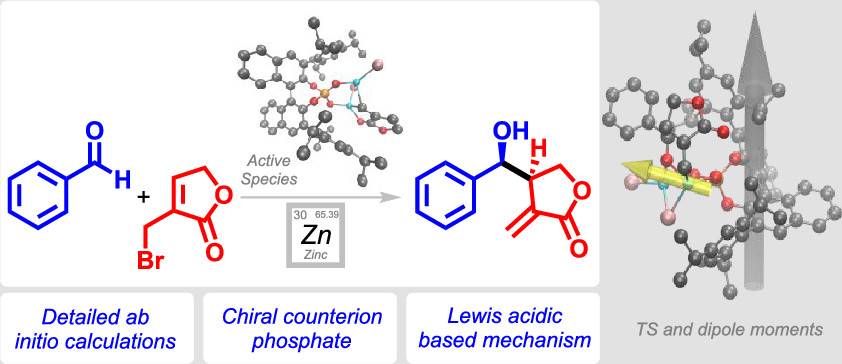
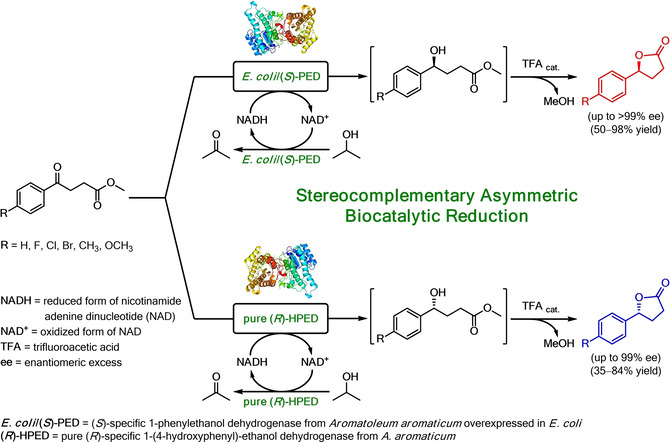

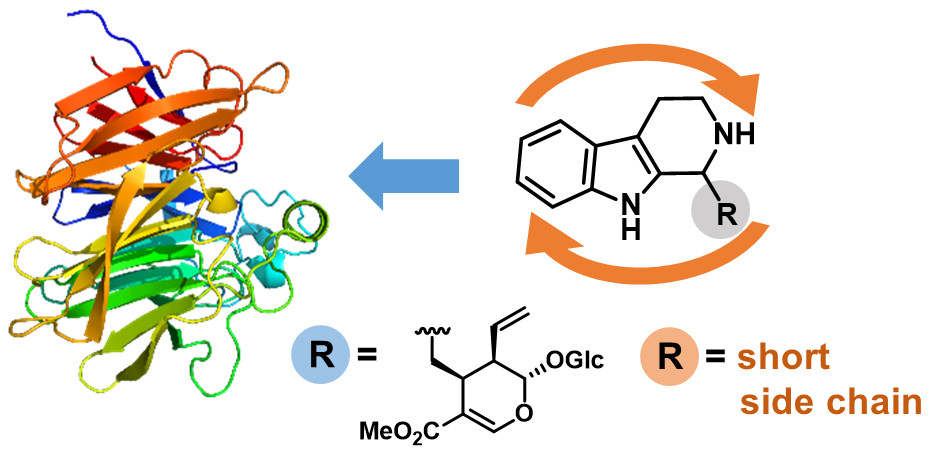
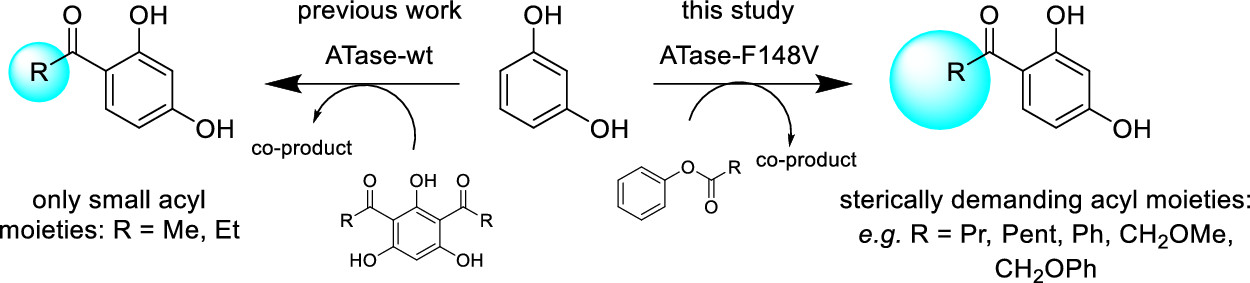
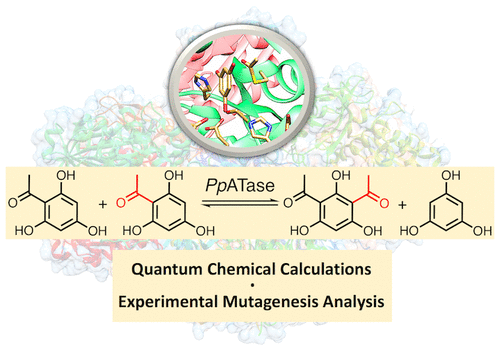
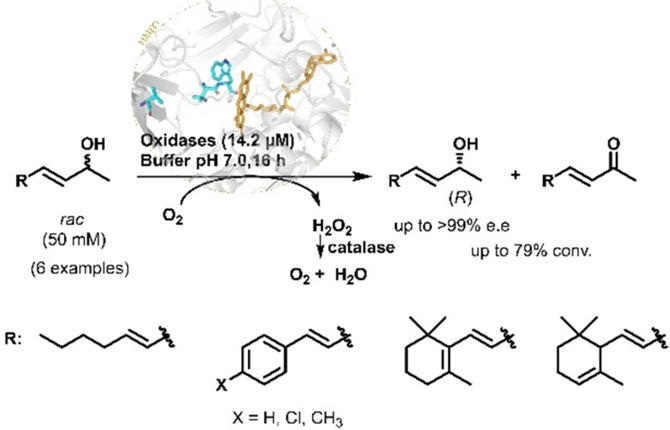
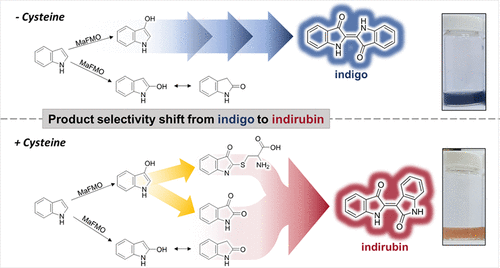



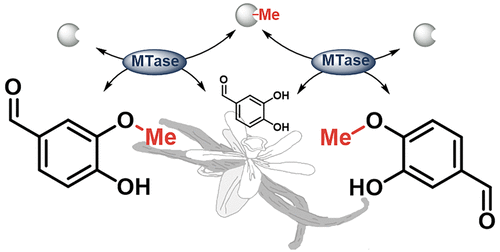
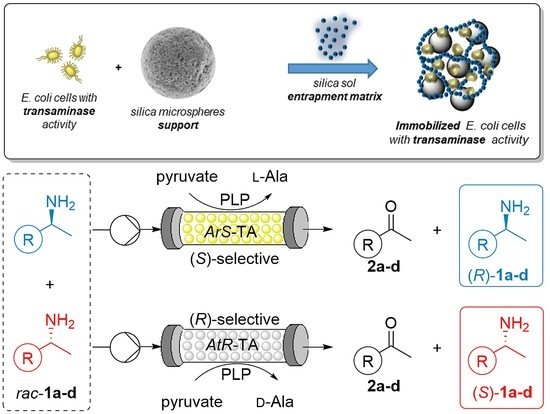

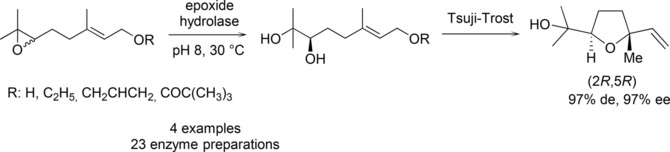
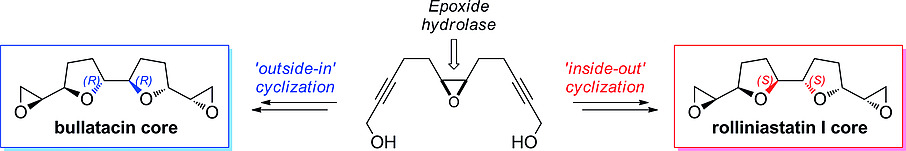
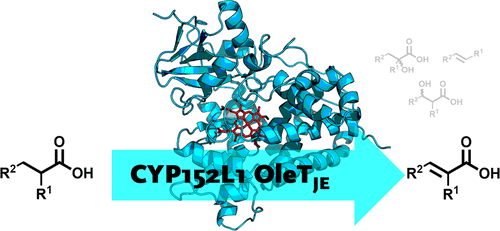
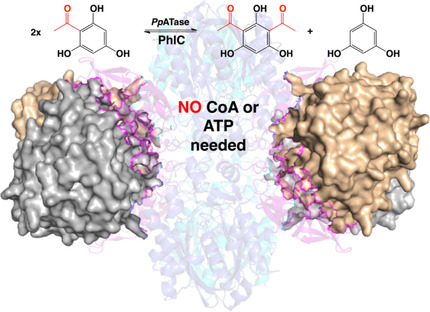
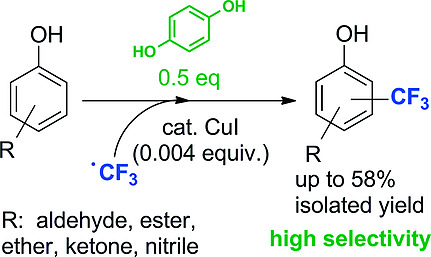
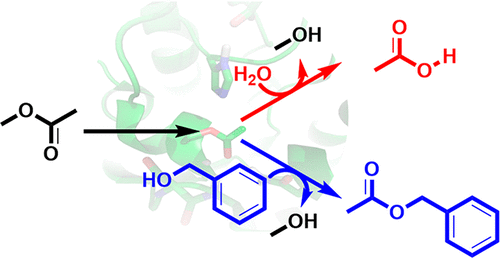


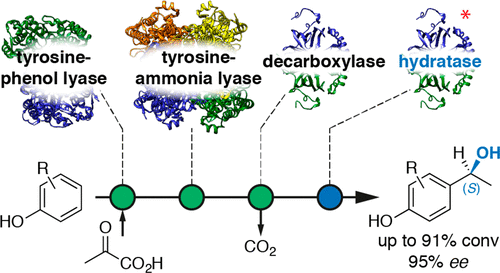

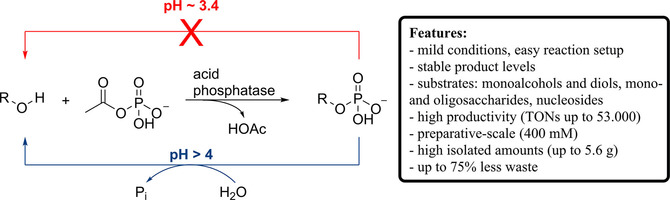

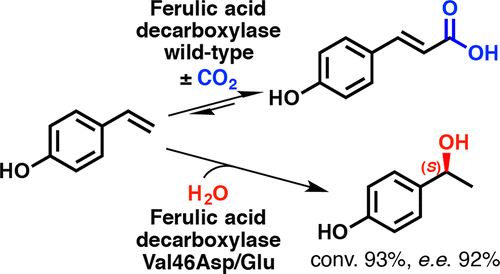


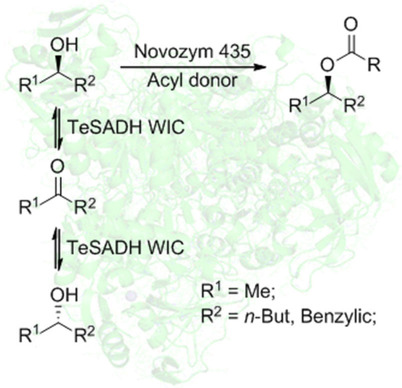
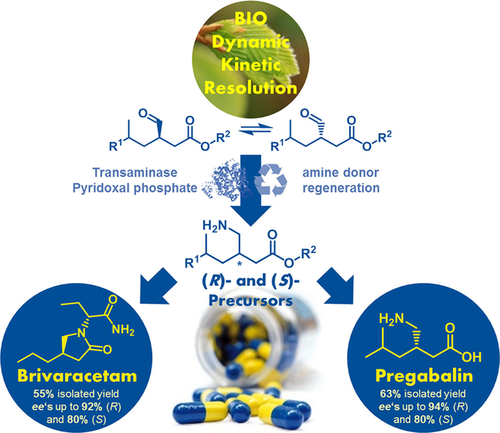
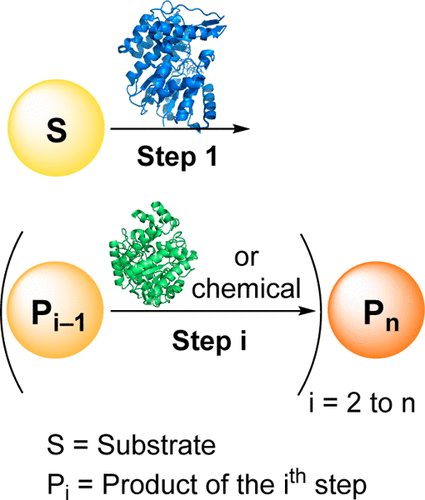
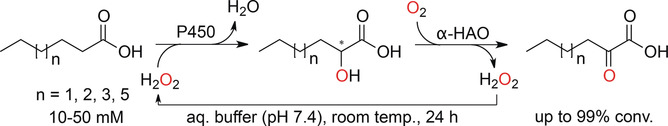
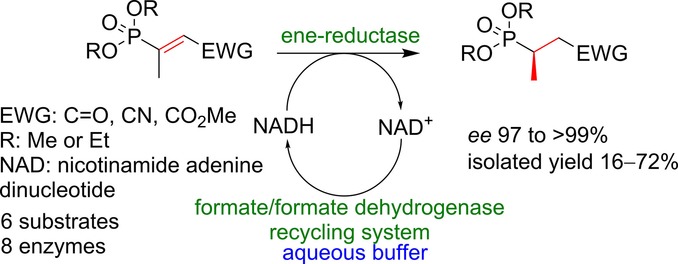
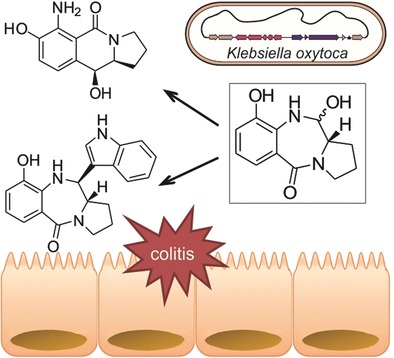

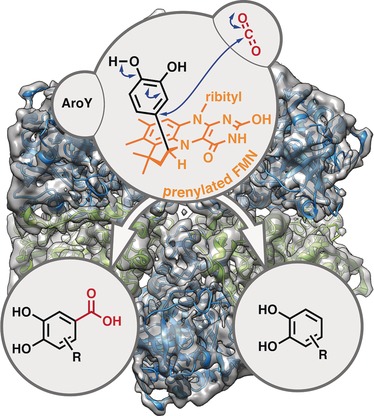

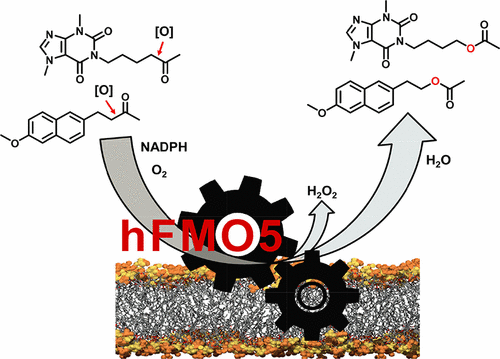
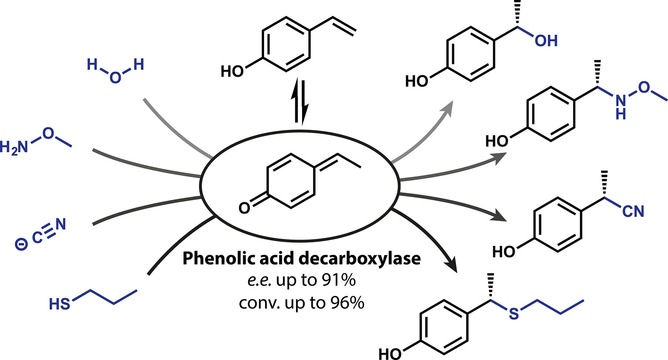
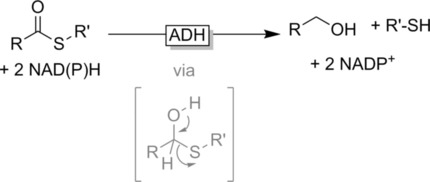
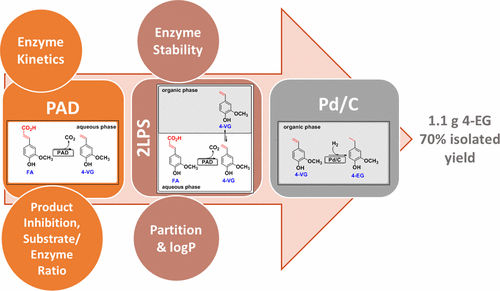
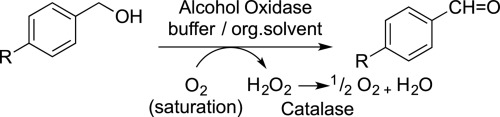
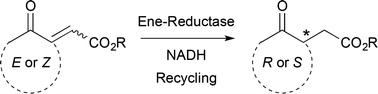

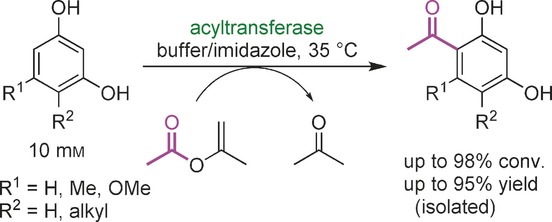

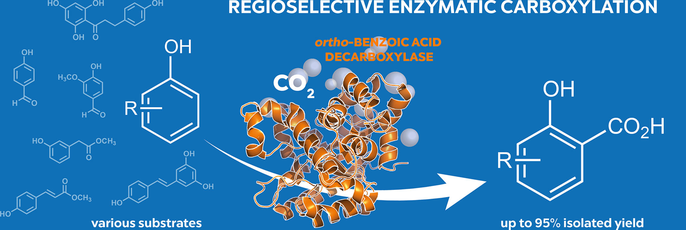
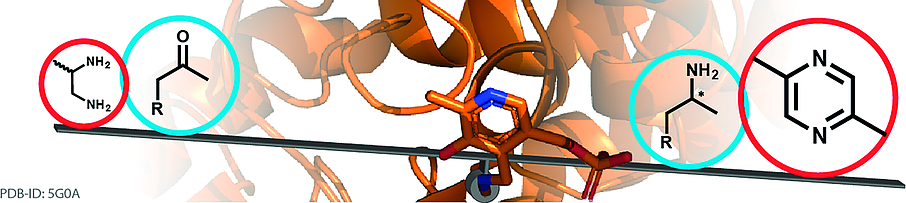
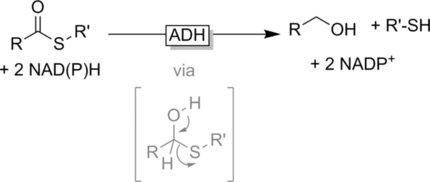
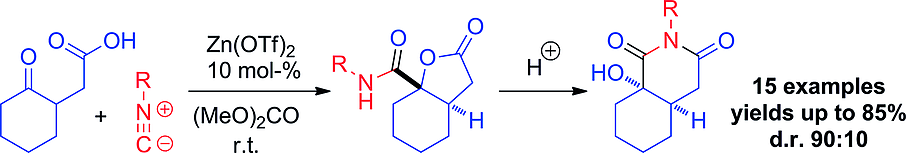
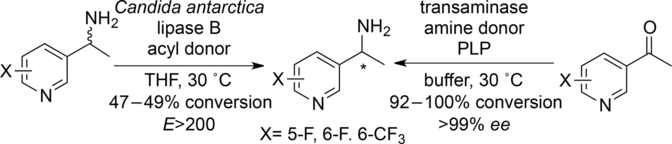
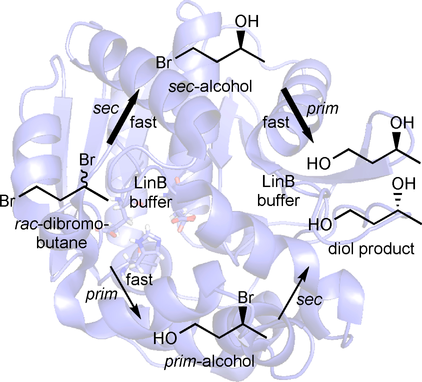
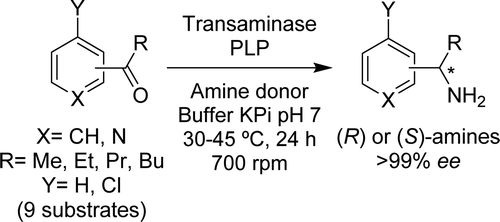

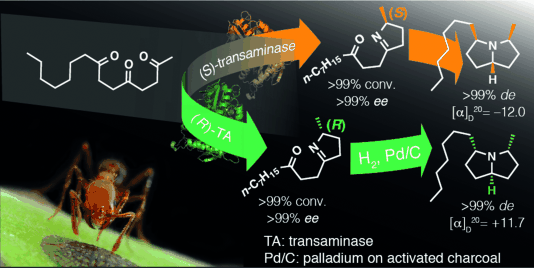
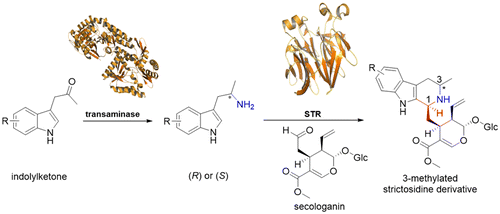

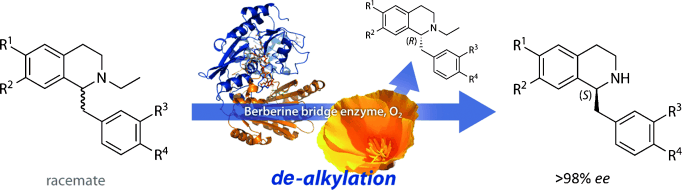
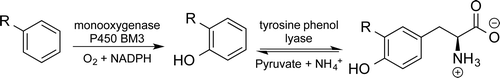
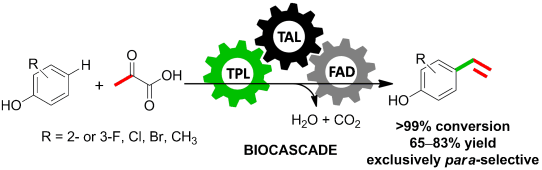
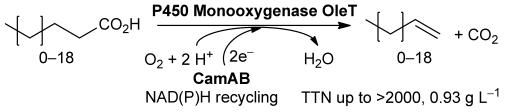
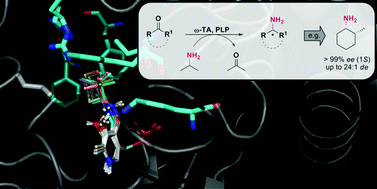
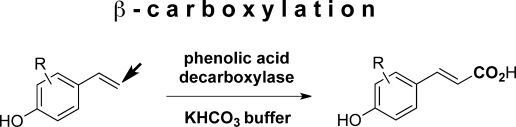







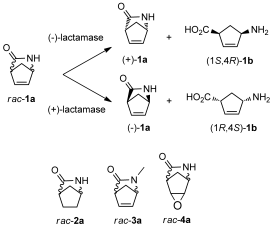
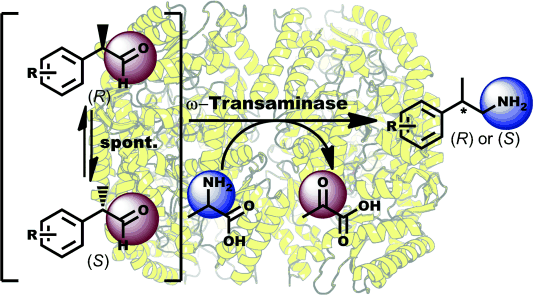
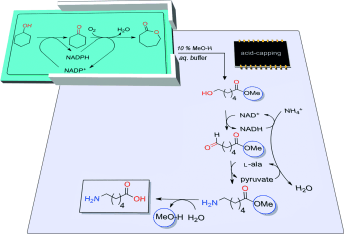

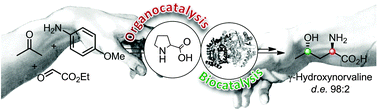
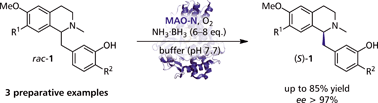
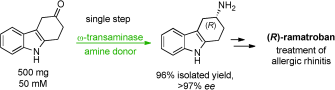
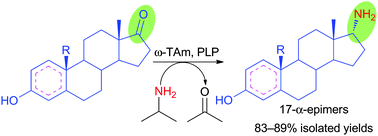
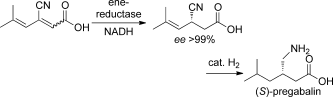

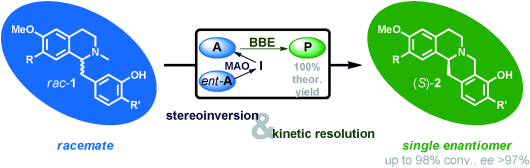
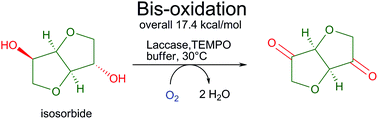
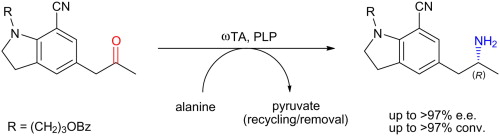

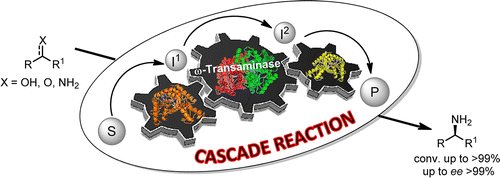

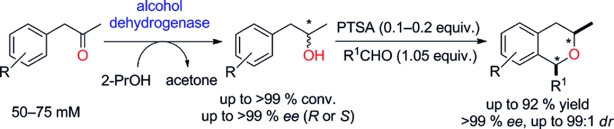
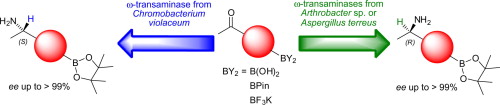
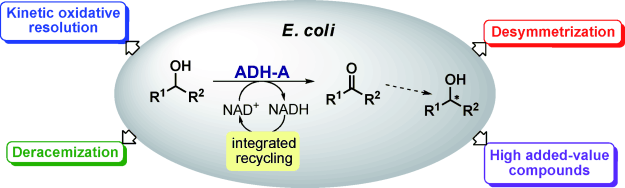
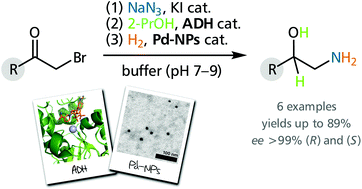
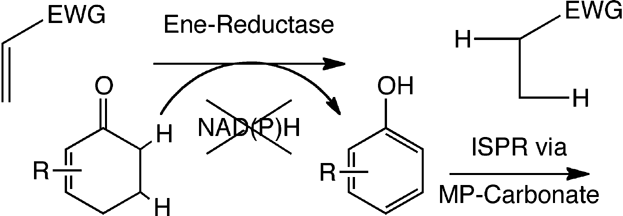
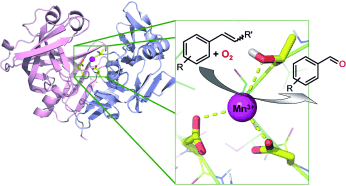
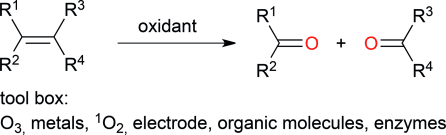
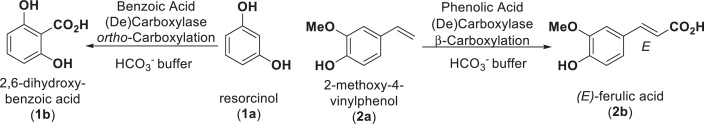
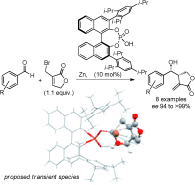

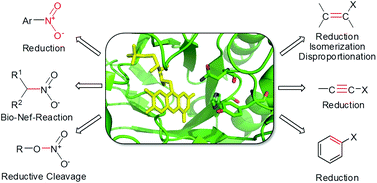
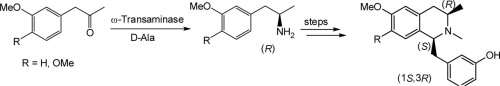
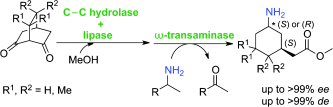

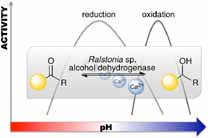


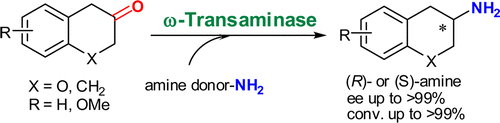


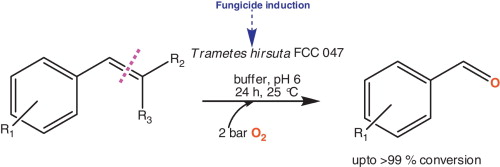
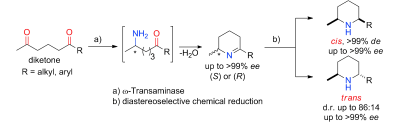
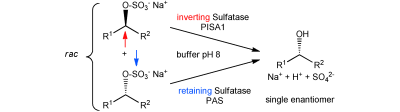
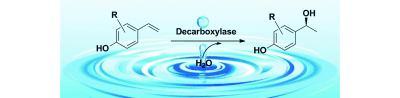
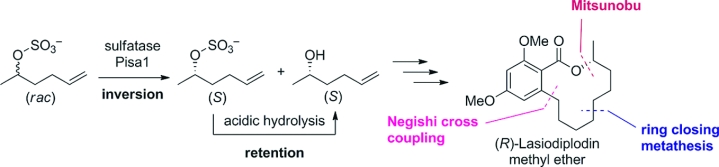
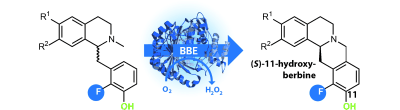

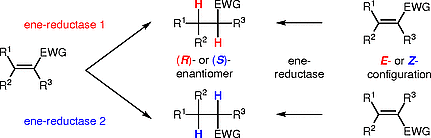
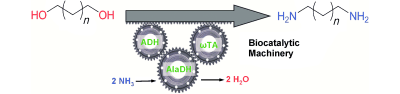
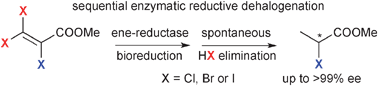

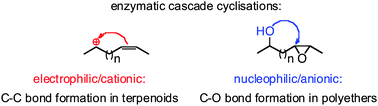
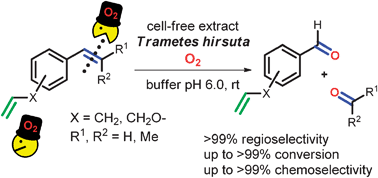
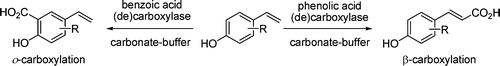
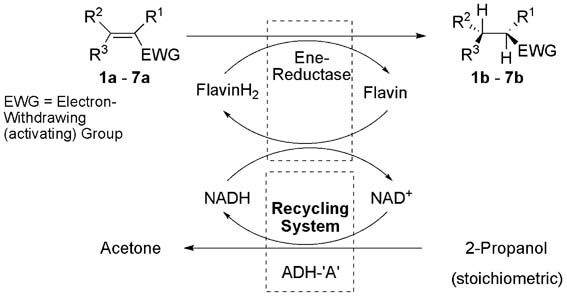
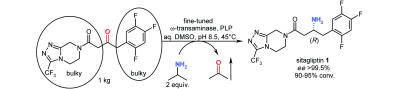
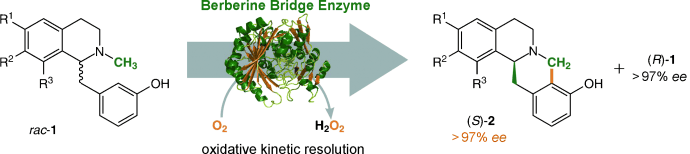

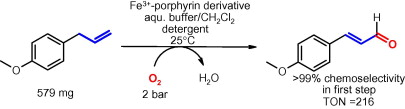
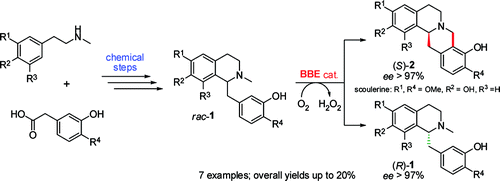
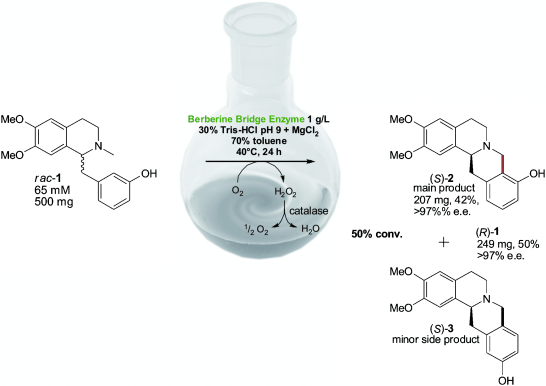
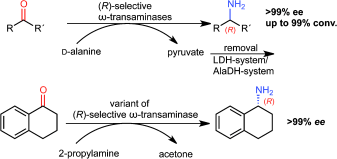
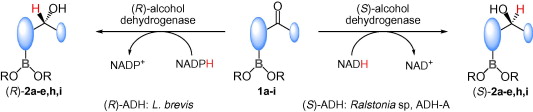
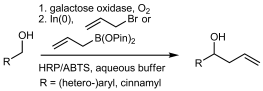
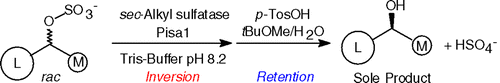


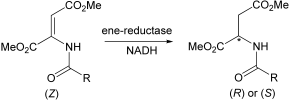

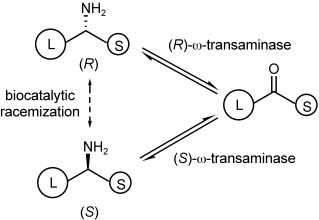

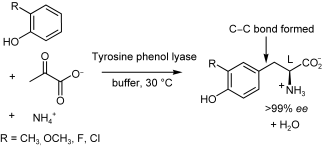
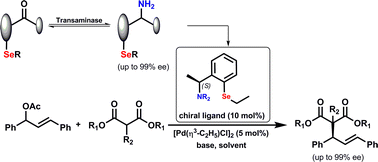

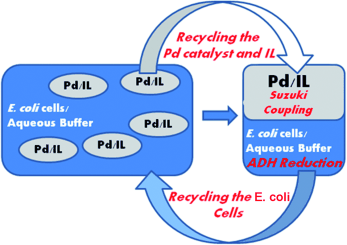
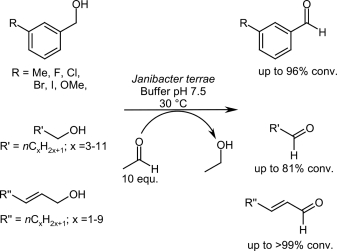
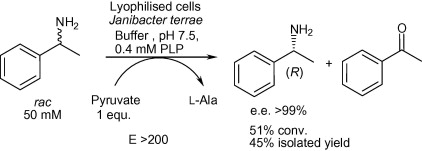
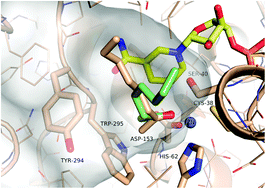
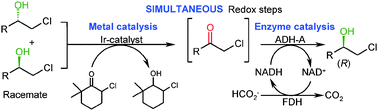

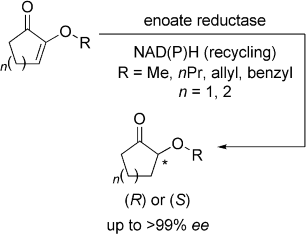
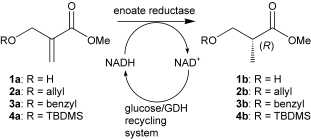
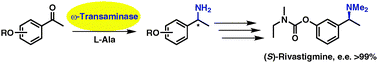


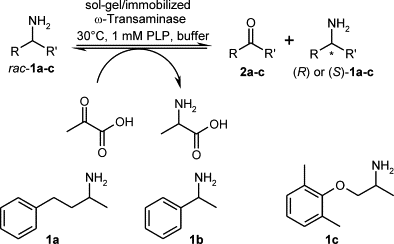
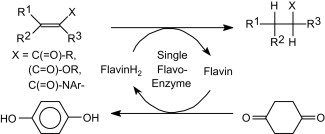
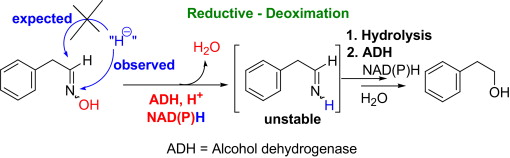

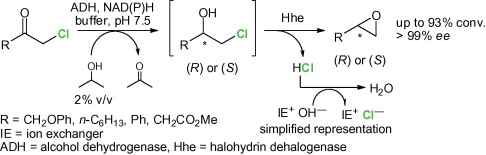

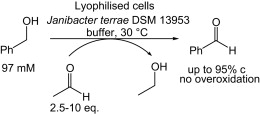
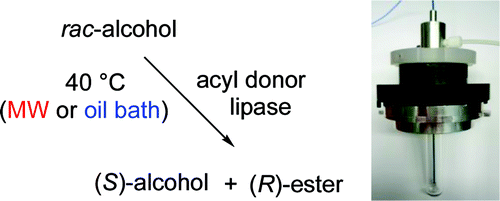
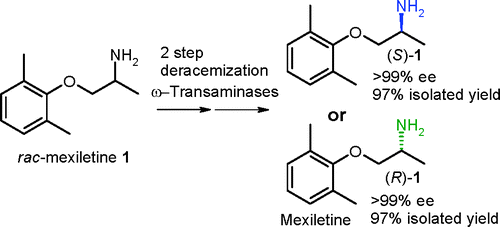
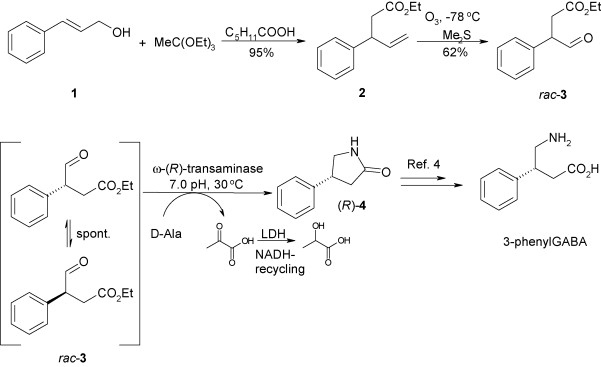
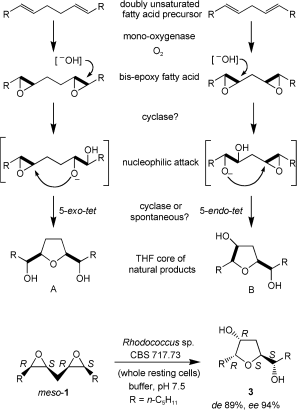


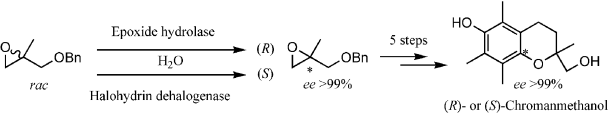





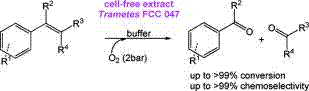
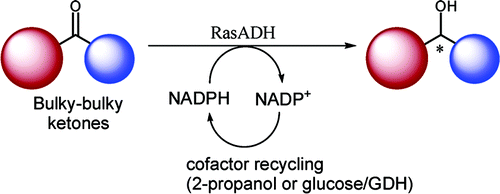
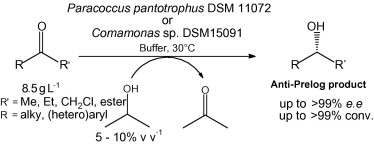
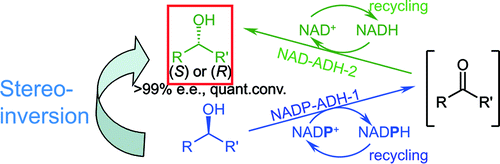


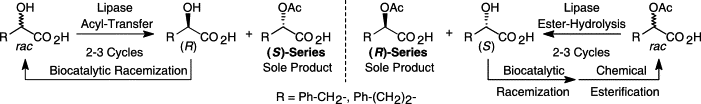
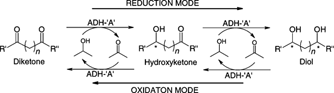


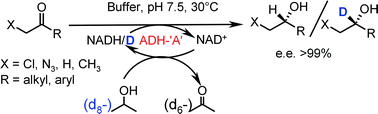
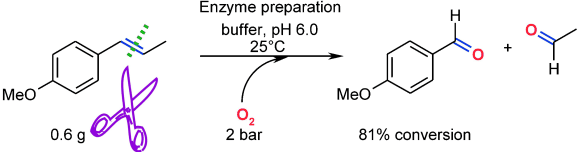





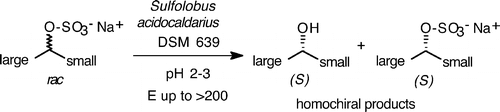
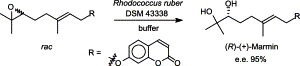
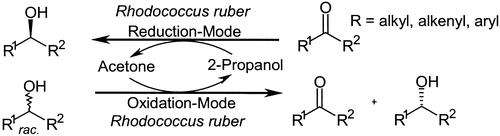
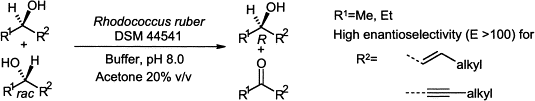
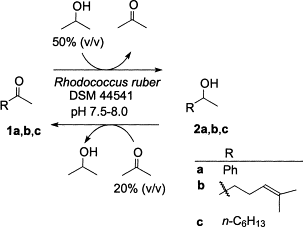


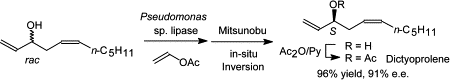
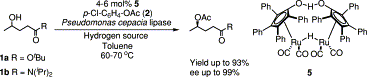
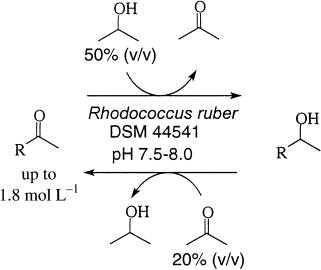


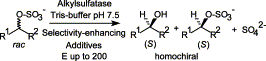


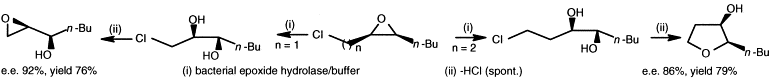
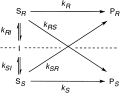

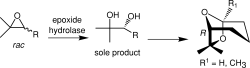
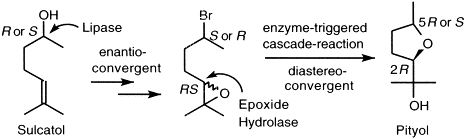
 .
.